Streamline Your ePRO Implementation: Top 5 Best Practices
Integrating new technology into your clinical research can seem overwhelming, but with the right approach, electronic Patient Reported Outcomes (ePRO) can be a worthwhile investment for gathering high-quality data directly from patients. To help you navigate this journey, we’ve distilled five essential best practices for getting started with ePRO. These tips are designed to be straightforward and effective, ensuring you harness the full potential of ePRO technology.
1. Start Simple
The key to successful ePRO implementation is to begin with a manageable scope and to ideally outsource services to a single ePRO provider, treating this as an opportunity to learn. Adhere to the best practices suggested by your provider, even including the frequency of notifications and the layout of reports. This approach reduces complexity and sets a solid foundation to build upon for future studies.
2. Start Early
Preparation is key. Kick off your ePRO project with ample time on your side. A visual timeline can be incredibly helpful here to navigate you through the process, outlining each phase of implementation, from planning and design to user acceptance testing (UAT) and go-live. An early start ensures you have the buffer to accommodate time to learn the processes without derailing your study milestones.
3. Identify Your Team Members
Assemble a dedicated ePRO project team early on. This team should include a main point of contact (POC) for the ePRO provider, representatives from Clinical Operations, Medical Lead, Health Outcomes Lead, Data Management Lead, Lead Clinical Research Associate (CRA), and your Contract Research Organization (CRO) if applicable. These individuals will be crucial for design discussions, status meetings, and making pivotal decisions about the ePRO implementation.
4. Use the ePRO Solution Before UAT
Familiarize yourself with the ePRO solution well before the User Acceptance Testing (UAT) phase. Engaging with the tool in advance helps inform design decisions, ensuring the platform meets your needs. Early exposure can significantly reduce the likelihood of last-minute changes during UAT, a time when flexibility is limited, and deadlines are fast approaching.
5. Minimize Changes After Go-Live
Post-launch adjustments may seem simple but can introduce significant challenges. Whether it’s adding a new ePRO tool, altering the schedule, or modifying protocols, each change requires careful management to avoid disrupting ongoing studies. Any alterations need retraining for sites and CRAs, may impact ethical committee or institutional review board documentation for participants, and could affect data reporting and exports. Therefore, it’s crucial to minimize changes after your ePRO system goes live to maintain study integrity and data consistency.
By following these five best practices, you’ll position your ePRO implementation for success, enhancing the efficiency and effectiveness of patient-reported data collection in your clinical trials. Remember, a thoughtful approach to implementation can make a significant difference in the long-term value and utility of your ePRO system.
In the landscape of clinical trials, electronic capture of patient-reported outcome (ePRO) data has emerged as a superior alternative to traditional paper-based methods. Electronic approaches offer a range of benefits, including improved data quality, reduced errors, and increased efficiency [Contemporary Clinical Trials Communications]. However, the provisioned device (PD) model, where trial sites manage and distribute dedicated devices to participants, comes with its own set of challenges and limitations.
Enter Bring Your Own Device (BYOD), a patient-centric strategy that allows participants to use their personal smartphones or tablets for ePRO data capture. BYOD offers a multitude of advantages that have the potential to revolutionize the eCOA landscape, enhancing patient engagement while simultaneously reducing the logistical and financial burdens associated with provisioned devices.
BYOD for trial participants
Firstly, BYOD prioritizes flexibility and convenience for trial participants. By enabling individuals to complete ePRO assessments on their own trusted devices, which are already seamlessly integrated into their daily lives, BYOD capitalizes on existing habits and behaviors. With the average smartphone user checking their device over 50 times per day, the familiarity and accessibility of personal devices have the potential to boost participant engagement and compliance, as participants clearly prefer it.
BYOD for sites & sponsors
Moreover, BYOD alleviates the logistical challenges and costs associated with provisioned devices. Trial sites are liberated from the burden of storing, tracking, and maintaining device inventories, allowing them to focus on their core responsibilities. Sponsors can realize significant financial savings by eliminating the need to purchase and distribute study-specific devices, which often go unused and wasted in the traditional PD model. With BYOD, sponsors only need to pay for the actual devices being used by participants, rather than an excess of provisioned devices that may never be deployed. This “pay as you go” approach to device provisioning, offered by forward-thinking eCOA providers like Castor, ensures that sponsors are not burdened with unnecessary costs.
Furthermore, trial sites, the key stakeholders in ePRO implementation, are increasingly recognizing the benefits of BYOD. A survey by Haenel et al. revealed that sites with prior BYOD experience tended to prefer it over PD, citing reduced logistical burden and increased participant convenience as major advantages [Contemporary Clinical Trials Communications].
Naturally, the question arises: is data captured via BYOD truly comparable to that obtained through provisioned devices or paper forms? Encouragingly, a growing body of evidence supports the measurement equivalence of BYOD ePRO data. A randomized study by Byrom et al. demonstrated that BYOD yielded comparable results to PD and paper modes across common ePRO response scale types in chronic pain patients [Value in Health]. Similarly, Hudgens et al. found strong measurement comparability between PD and BYOD for daily and weekly assessments in COPD patients over a 15-day period [Journal of Patient-Reported Outcomes].
Taking a flexible approach
At Castor, we firmly believe that BYOD represents the future of eCOA. However, we also understand that sponsors may have varying requirements and preferences. That’s why we offer a flexible approach, supporting both BYOD and PD models. Our “pay as you go” provisioning ensures that sponsors only pay for devices that are actually shipped to patients, eliminating the wasteful expense of unused provisioned devices that plagues many traditional eCOA providers.
As the clinical trials industry strives to enhance patient-centricity and operational efficiency, BYOD emerges as a compelling solution. By harnessing the power of personal devices and leveraging innovative eCOA platforms like Castor’s, we can create a more engaging, streamlined, and cost-effective eCOA process that benefits all stakeholders. The evidence is clear: BYOD offers measurement equivalence to traditional modes while providing tangible advantages for participants, sites, and sponsors. It’s time to embrace this patient-centric approach and usher in a new era of efficiency and engagement in clinical trials.
For more information, follow us on LinkedIn: Castor | Derk Arts
Our recent webinar featuring PRO expert Ari Gnanasakthy, RTI Health Solutions, and Derk Arts, CEO & Founder, Castor, sheds light on the complexities and advancements in measuring PROs, drug tolerability, and quality of life in cancer trials. Here, we distill the insights shared and explore the implications for future research.
Cancer studies are not what they used to be
Derk and Ari kicked off their conversation by discussing how cancer research is getting more and more complex and with this ever-evolving landscape, a focus on patient-reported outcomes (PROs) has never been more critical. With the emergence of innovative treatments such as plasma-derived therapies, CAR-T therapies, reductions in chemotherapy cycles, and a surge in novel oral medications, the shift towards more personalized and targeted therapies underscores the necessity to move beyond traditional PRO methodologies.
In this new era, a more patient-centric approach is needed, ensuring that treatment strategies not only combat the disease effectively but also offer real-time insights into treatment efficacy, patient well-being, tolerability, and quality of life (QoL).
“Let’s stop talking about patient reported outcomes, instead let’s talk about the patient experience and the ways we measure QoL within all the nuances of cancer trials.“
– Ari Gnanasakthy, RTI Health Solutions
Navigating the regulatory landscape
The webinar delved into the changing regulatory landscape for cancer trials, particularly for measuring PROs. As treatments like CAR-T cell therapy become more prevalent, capturing the authentic patient journey has become more crucial for understanding both efficacy and safety. Regulatory bodies are increasingly emphasizing the importance of real-world evidence, pushing for a more comprehensive view of treatment impacts. This shift necessitates meticulous planning in data collection, not only to meet regulatory approval requirements but also to address the downstream needs of payers. Having this dual focus ensures that the collected information serves both immediate regulatory purposes and future payer evaluations, facilitating a smoother transition from clinical approval to market access. With the rise of personalized and targeted therapies, the reliance on outdated PRO methodologies is no longer viable. Clinical trial design needs to reflect a balance between scientific rigor and patient-centricity, ensuring research outcomes are both scientifically valid and meaningful to patients’ lives.
Who says the drug is tolerable?
The Patient. A critical aspect of the discussion between Ari and Derk centered on the concept of measuring treatment tolerability and QoL within cancer research. The conversation highlighted a need to shift perspectives, with emphasis on understanding how tolerability influences patient adherence and overall treatment outcomes. Ari emphasized the importance of viewing tolerability through a comprehensive lens and throughout treatment cycles.
“Tolerability is not something that can be measured at the beginning of each cycle when patients are reasonably healthy. Tolerability is something that needs to be measured when patients are having issues and when the drug is at peak, during cycles…probably on a weekly basis.”
– Ari Gnanasakthy, RTI Health Solutions
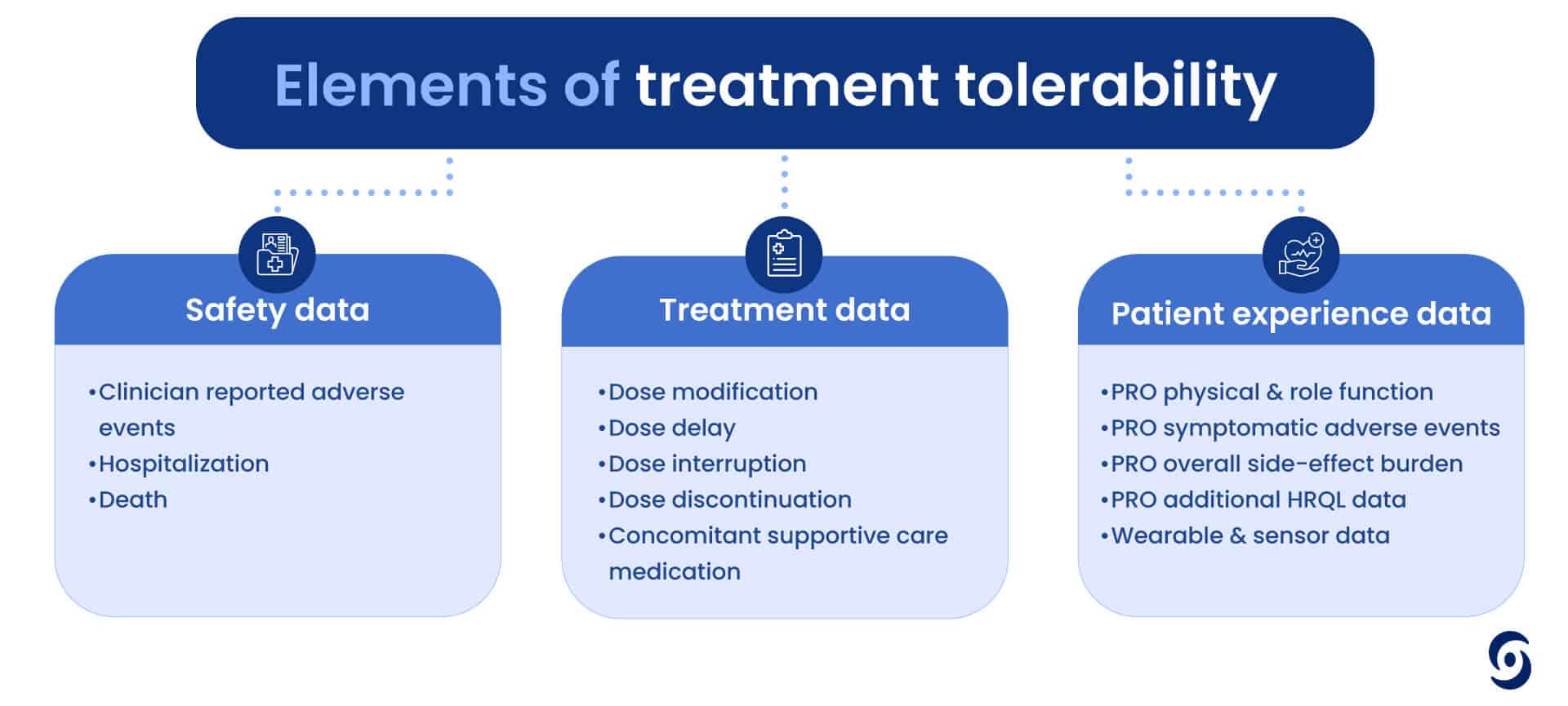
But how do we consistently measure something so subjective in a scientific way? Tolerability is not just one measure but a number of elements that need to be considered, including safety data, treatment data and PROs (like physical and role function, side effect burden or even hair loss). It is clear we need to consider both the physical and psychological nuances of patient experiences when assessing treatment tolerability.
What does the future hold for ePRO?
Finally, Derk and Ari spent time looking ahead at how the role of technology and ePRO in cancer research is expected to expand. Wearables, BYOD (Bring Your Own Device) strategies and advanced scheduling for data collection will play pivotal roles, enabling real world data collection that reflects the true impact of cancer treatment on patients’ lives.
This digital-first approach is anticipated to improve data quality and drive more patient engagement. By engaging patients through their own smartphones and leveraging technology they are familiar with, clinical trials can more accurately measure the true patient experience, thereby improving participation and adherence. Additionally, ePRO technology offers solutions to challenges around data integrity and overcoming the “parking lot effect,” where responses may be influenced by the immediate environment of data collection. This evolution marks a pivotal step towards more patient-centric research and ultimately better outcomes.
“It’s now clear that we’re dealing with three layers of data. Initially, we have the core data that has been the focus for the past two decades. Added to this are scheduled QoL and tolerability data. The final layer encompasses wearable and ad hoc data, enriching our understanding of tolerability. This stratified approach presents a significant opportunity to enhance patient outcomes.”
– Derk Arts, Castor Founder & CEO
And, from the sponsor side, it’s imperative to consider the influence of Quality of Life (QoL) and tolerability measures on the drug’s value proposition. For sponsors, overlooking this aspect could lead to a significant gap in their value proposition.
The single line item from the FACT-G questionnaire, GP5, which asks patients if they are “bothered by side effects of treatment,” has demonstrated a strong predictive value for both patient dropout and disease progression when patients report being troubled by side effects. This underscores the importance of integrating patient-centric measures into clinical trial design and evaluation to enhance patient retention and treatment outcomes.
Getting started with Castor ePRO
To deepen the understanding of ePRO technology for sites and sponsors, and to ensure fast study builds, Castor welcomes study teams to explore our ePRO platform by offering early access, and allowing them to personally navigate the patient experience. Our comprehensive onboarding process and dedicated customer support play a pivotal role in this commitment. Additionally, we provide the option to work with synthetic data upfront, offering a proactive approach to risk mitigation.
At the heart of our platform is a patient-centric approach, designed to significantly improve the trial experience for participants. By prioritizing the patient perspective, we aim to enhance engagement and collect valuable real-world data. This data is crucial for gaining insights into treatment effectiveness, patient well-being, tolerability, and quality of life, in the ever changing landscape of cancer research.
Over recent decades, we’ve witnessed a pivotal shift in the global healthcare landscape. As chronic conditions have become more prevalent, nations worldwide have been reevaluating their healthcare delivery models, moving away from traditional fee-for-service models towards a more outcomes-focused approach. Both governments and private payers are now pondering a critical question: “How can we reimburse healthcare providers based on the positive outcomes for patients, rather than just for the services they provide?”
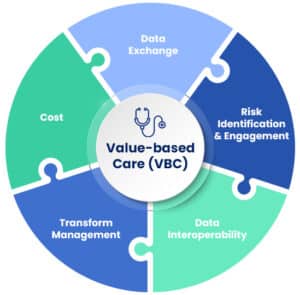
Value-Based Care
Value-Based Care (VBC) programs represent a transformative approach in the healthcare industry, aimed at improving patient outcomes, enhancing care quality, and reducing healthcare costs. Unlike traditional fee-for-service models, which reimburse healthcare providers based on the quantity of care services delivered, VBC focuses on rewarding providers for the quality and effectiveness of care they provide to their patients. This model aligns financial incentives with patient outcomes, encouraging healthcare providers to offer care that is both efficient and tailored to the individual needs of patients.
The impact of VBC on the healthcare ecosystem is multifaceted. Firstly, it prompts a shift in the focus of healthcare delivery from treatment to prevention and patient wellness. By incentivizing positive patient outcomes, VBC encourages providers to invest more in preventive care measures, chronic disease management, and holistic approaches to patient health. This can lead to a decrease in hospital readmissions, fewer unnecessary medical procedures, and overall, a more sustainable healthcare system.
Secondly, VBC fosters collaboration among healthcare providers. Since the model rewards the improvement of patient outcomes across the continuum of care, it encourages different providers to work together more closely. Hospitals, primary care physicians, specialists, and even non-medical support services are motivated to share information and coordinate care more effectively, ensuring that patients receive the right care at the right time.
Patient-Reported Outcomes (PROs) play a crucial role in the success of VBC programs. PROs are direct reports from patients about how they feel in relation to a health condition and its therapy, without interpretation of the patient’s response by a clinician or anyone else.
This information is invaluable because it provides insights into the patient’s perspective on their health status, quality of life, and the effectiveness of treatments.
In the context of VBC, PROs help to ensure that care delivery is genuinely patient-centered, enabling healthcare providers to tailor their interventions to meet the specific needs and preferences of each patient.
PROs contribute to the measurement and assessment of healthcare outcomes, which is central to VBC. By incorporating PROs into their assessment criteria, VBC programs can use this data to evaluate the effectiveness of care from the patient’s viewpoint. This ensures that healthcare interventions not only achieve clinical objectives but also improve the quality of life for patients. Consequently, PROs are essential for the continuous improvement of healthcare services, guiding providers towards interventions that offer the greatest benefit to patients’ health and well-being.
PROs in Clinical Trials
When life sciences companies present data from clinical trials that include PROs, they provide a more comprehensive picture of a product’s benefits. This evidence is particularly persuasive to payers and healthcare providers, who are increasingly looking for treatments that offer meaningful improvements in patients’ lives, not just clinical metrics. For payers, such information supports decision-making related to coverage and reimbursement, as it aligns with the shift towards outcomes-based reimbursement models. Products that demonstrate a positive impact on patients’ quality of life are more likely to be covered and recommended within these frameworks, ensuring broader access for patients.
Similarly, healthcare providers, who are integral to the VBC ecosystem, are more likely to prescribe products that have been shown to enhance patient outcomes and satisfaction. In the context of outcomes-based care, providers are rewarded for delivering high-quality, patient-centered care, which includes prescribing treatments that patients are more likely to adhere to because of their favorable impact on quality of life. Therefore, clinical trial data enriched with PROs can significantly influence prescribing behaviors by highlighting the patient-perceived benefits of a treatment.
The inclusion of PROs in clinical trials signals to both payers and providers that a life sciences company is committed to understanding and addressing the holistic needs of patients. This can strengthen the company’s position in negotiations with payers and foster trust among healthcare providers, facilitating the successful adoption of new products.
PROs bridge the gap between clinical efficacy and patient experience, enabling life sciences companies to better meet the demands of the evolving healthcare landscape where the patient’s voice is increasingly central to care decisions.
Leveraging PROs in clinical trials is not just about enhancing the evidence base for new medical products; it’s about aligning these products with the fundamental principles of VBC and ensuring they meet the real-world needs of patients, payers, and providers. In doing so, life sciences companies can enhance the marketability of their products, ensuring they are well-positioned for successful adoption, coverage, and use in a healthcare environment that values and rewards meaningful improvements in patient health and quality of life.
Reflecting back on the SCOPE 2024 conference held in Orlando, FL last week, it became evident that Artificial Intelligence (AI) is no longer just a buzzword but a pivotal force driving innovation across drug development processes. From protocol design to submission readiness, and internal operational efficiencies, Big Pharma is harnessing AI’s potential to streamline operations and accelerate the journey of drugs to patients. However, as the industry navigates through the exploratory phase of AI applications, a critical question arises: Should companies work together and collaborate to speed up innovation through standardization and synergy, or does a healthy dose of competition serve as the catalyst needed for rapid advancements? Here we delve into the debate, informed by insights from panel discussions at SCOPE 2024 on generative AI in clinical research, focusing on technology and data challenges.
AI Applications in Big Pharma
AI’s footprint in Big Pharma is expanding, with several companies exploring identical use-cases in data science, clinical operations, regulatory affairs, and beyond. Common applications include protocol design, ensuring submission readiness, deploying internal chatbots for policy queries, and creating co-pilots for document generation. Companies like Pfizer, Lilly, and AbbVie have shared how they are leveraging AI to not only enhance efficiency but also to facilitate a more coherent and consistent approach to drug development. Specific use-cases discussed at SCOPE 2024 included:
Automating document generation to streamline, standardize and create comprehensive submission packages
AI powered summarization of complaints, acting as ‘the human eagle eye’ and using AI to identify potential root causes for these complaints
The Case for Collaboration
Collaboration in AI could pave the way for standardization and synergy, leading to universally accepted practices and platforms that could significantly reduce redundant efforts and costs. By sharing knowledge, resources, and data, companies can avoid duplicating work and instead focus on scaling successful AI applications more efficiently. However, the path to collaboration is fraught with challenges, including aligning on common goals, intellectual property concerns, and the time-intensive nature of establishing industry-wide standards.
The Case for Competition
On the other side of the debate, competition is seen as a driving force for innovation, much like the historical example of the Space Race, which catalyzed unprecedented advancement in technology and space exploration. The desire to outperform rivals can lead to creative new solutions, accelerating the development and implementation of AI applications. Competition encourages companies to push the boundaries of what’s possible, leading to breakthroughs that might not occur in a collaborative environment. However, this approach can lead to scalability issues and the potential for fragmented systems that may hinder rather than help the overall goal of bringing drugs to patients more quickly.
The Path Forward
The debate between fostering collaboration and encouraging competition in AI innovation within Big Pharma is complex, with valid arguments on both sides. While collaboration offers the promise of standardization and shared progress, competition drives rapid innovation and creative problem-solving. Ultimately, the path forward may not be an either/or scenario but a balanced approach that leverages the strengths of both strategies to accelerate the delivery of life-saving drugs to patients.
Building a compelling business case for AI involves identifying clear ROI and focusing on solving tangible business problems. For instance, IQVIA’s approach to AI categorizes applications into language and data, aiming to enhance digital protocol design and predict patient enrollment more accurately. Such strategic focuses not only clarify the value proposition of AI but also ensure that technological investments are directly aligned with business objectives.
What’s next?
As the pharmaceutical industry continues to evolve with AI, engaging in open dialogue and sharing best practices will be crucial. Stakeholders are encouraged to contribute their insights and experiences, fostering an environment where innovation can flourish, guided by both collaborative spirit and competitive drive. The journey of AI in pharma is just beginning, and together, we can shape its trajectory for getting much needed treatments to patients faster.
For more information, follow us on LinkedIn: Castor | Derk Arts
An avalanche of data from so many real-world sources is available today, with the promise of more data generated daily—so why not use it?
Real-World Evidence is evolving before our eyes
Real-world evidence (RWE) is a hot topic with the FDA and those submitting to the FDA, and it is a topic that promises to grow in interest and relevance. Since the Cures Act of 2016, the FDA has been looking for and finding reasons to pay attention to RWE made up of Real-world data (RWD). With increasing urgency, RWE has been finding its way into regulatory submissions for new products.
RWD is rising to the authoritative level of randomized clinical trials (RCT)—the standard-bearer of definitive evidence. RCTs are designed and administered to produce the results needed to meet stringent clinical criteria, while RWD generally collects data to support something other than a clinical trial. RWD is if anything, an afterthought when it comes to supporting clinical trials. But with increasing amounts of data becoming more available every day, it was only a matter of time before the rigor and methodologies that power RCTs get designed into the RWD that is churning out day in and day out. The RWE generated by RWD will not always be only an afterthought.
In this article, we want to show the gaps between how data is collected in RWE and RCT and how RWE is quickly finding ways to catch up: from applying rigor to RWE by reusing data models and repurposing methodologies from RCTS to creating fit-for-purpose methodologies for post-market surveillance.
While the FDA looks at RWE as a part of the evidence package, the agency does not allow RWE to meet a lower threshold of evidence compared to studies using RCTs. In fact, not all RWE was accepted as the evidence it was intended to provide. In a review of FDA documentation, Papura et al. observed the FDA citing the use of the three categories of evidence noted above (substantial, primary, supportive) to approve new license applications. They also noted that the RWE did not always rise to the level of supporting the FDA’s benefit-risk assessment framework.
To meet the exacting needs and frameworks of the FDA, it makes sense to look at adopting the science behind RCT. In particular, the methodologies that make RCTs must be adapted to produce fit-for-purpose RWD (that is, data that meets the more stringent criteria found in an RCT). For instance, RWD must meet the same relevance and reliability standards used for RCTs.
Relevance: The data should represent the intended population. The data should capture exposure, and confounders should be captured and measured.
Reliability: Ensure accuracy, completeness, and consistency
Identifying and adjusting for demographics, socioeconomic and insurance status, disease severity, comorbidities, and other confounding factors are also important. One of the important questions facing the use of RWE is the question of systematic bias.
Are the patients in the dataset representative of the population of interest?
Are critical data fields representing exposures, covariates, and outcomes present? If not, are these variables able to be algorithmically derived using data fields that are present?
If more than one data source is required, are data fields present that permit accurate linking at the patient level?
Are there sufficient persons and follow-up time in the data source to demonstrate the expected treatment effect, including adequate capture of potential safety events?
A real-world dataset is relevant if it is robust and representative of the population of interest
– Center for Drug Evaluation and Research
RWE plays a bigger role in post-market clinical follow-up (PMCF)
The FDA Adverse Event Reporting System (FAERS) is one avenue for monitoring the PMCF experience with a drug. MedWatch is another program that allows for the voluntary reporting of serious reactions and problems with medical products. Clinical data registries are another important set of tools for tracking the details of disease or pathology progress. In addition to these and other programs, RWE is playing an increasingly important role.
Real-world evidence is traditionally used for PMCF studies. The use of RWE in postmarket surveillance adds richness to the data collected from clinical trials. Swift et al., in their look at the innovative uses of RWE, noted potential deficiencies of RCT. In particular, some inferences that arise from RCT can limit the ability to individualize for specific clinical scenarios in real-world medical practices. So planning to include RWE helps expand the application of evidence-based medicine.
The use of RWE for post-market surveillance is already a natural fit and becomes even more relevant as RWE is fit-for-purpose for the many stages of a therapy’s life cycle.
Tokenization and RWE
Tokenization takes RWE even further by making data available on a broader scale. Tokenization uses a unique, anonymized identifier (a “token”) to track a patient’s information without linking that information to the patient’s identity. Tokenization allows another researcher to link external data sources to the original study with a different (or adjacent) set of questions that examine the data from a different vantage point. For instance, according to Deborah Borfitz, writing in Clinical Research News, a study that used “time to next treatment” as an endpoint could be reviewed again by examining claims data, which could identify the time the participant was on the intervention. Tokenization allows creative thinking around the kinds of questions that can be asked of data.
Tokenization of RWE will prove useful in broadening the data sets available in the post-market surveillance phase, creating new avenues for verifying efficacy. We can expect faster and more efficient analysis as the tokenization of RWE draws from a variety of data sources, including EHR, claims data, and even social media posts. Results may include faster responses to safety concerns and better risk management strategies.
How Castor can help your real-world data capture strategies
RWE deserves to move out of the domain of afterthought. It rightly deserves a front and center seat in the study pipeline, which is precisely what the FDA and other regulatory bodies are eagerly pushing for. Beyond post-market surveillance, FDA considers RWE studies as part of the evidence package for submissions seeking authorization to market new medical products, including new drug applications (NDAs) and biologics license applications (BLAs).
Castor can help in the creation, collection, and analysis of RWE. Our experience with mobile apps, eConsent, and wearables can increase recruitment rates (by 50-65%) and patient retention (40% higher), which results in more and better RWE over time.
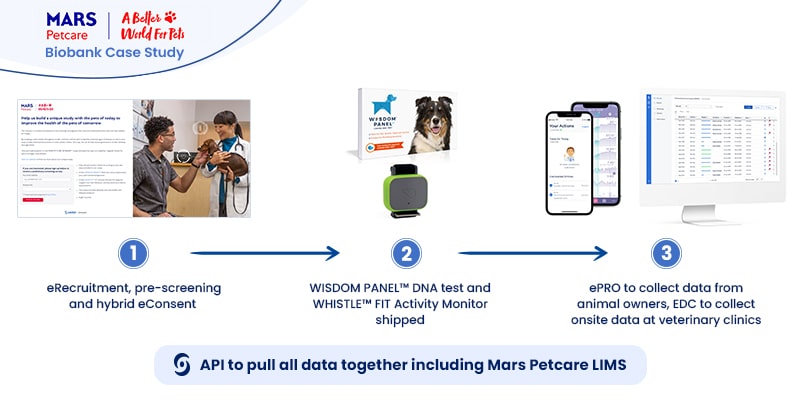
The MARS Petcare Biobank is an ambitious large-scale, longitudinal study to collect health and disease state information from 20,000 pets (10,000 cats/10,000 dogs) over ten years. The real-world, open-access data will be available for researchers, with raw genome sequences to be released as they become available. The study team was well aware of the enormous amount of information they would handle by collecting data from 20,000 wearables.Mars Petcare turned to Castor to help collect and process the tsunami of real-world data from tracking 20,000 pets in more than 50 sites for ten years. Real-world data would be gathered from WISDOM PANEL™ DNA tests to determine the pet’s breed background and the WHISTLE FIT™ activity monitor—both shipped to the pet owner’s home.Castor electronic data capture (EDC)—supported by the use of the RESTful API—helped to quickly capture and collect trial data produced by the many wearables and seamlessly integrate that data into the study. Castor products like eConsent and ePro helped speed initial phases of the study:
eConsent, by creating hybrid eRecruitment methods (online landing page & pre-screening surveys delivered online or in-person on paper).
ePro greatly simplified the participant-reported outcomes, which
EDC would collect onsite and in a single onsite repository
The informed consent process for clinical trials is often misconstrued as a single event—a signature hastily scribbled on a piece of paper. This is far from what is required by the ethical committees that work alongside regulatory bodies, like institutional review boards (IRBs). Instead, IRBs outline an ongoing and involved process that ensures participants know exactly what they are getting into. Electronic consent (eConsent) platforms advance the consent process by making content more comprehensive, accessible, and digestible for participants.
Here we’ll cover how eConsent benefits trials and aligns with IRB ethical commitments, how to prepare your IRB submission for approval, and the future impact of large language models on the informed consent process.
Informed consent is more than just a signature
Ethical committees are the administrative bodies that protect research participants’ rights, privacy, and welfare. They review informed consent processes to ensure patients understand the benefits and risks of participating in a clinical trial. In the US, these FDA-mandated groups are called IRBs; elsewhere, they are known as independent ethics committees, ethical review boards, or research ethics boards.
Informed consent is an important component of an IRB submission but is more than just a signature.
Providing a patient with all the information they need to make an informed decision
Allowing opportunities to test a patient’s understanding and for patients to ask questions
Continuing patient updates about impacts on their safety and wellbeing
In other words, informed consent must be holistic, interactive, and ongoing.
eConsent is more than just digital paper
The FDA encourages using eConsent because it provides additional opportunities for patients to connect with trial material and to stay updated throughout the process.
When properly designed, eConsent provides participants with alternative ways to view and process informed consent material outside a clinical setting, new possibilities for assessing knowledge and asking questions, options for participants to stay updated on pertinent information, and a cleanly-tiered presentation of information to satisfy IRBs without confusing participants.
Benefits of choosing eConsent
In addition to the consent process,eConsent platforms often include recruitment, screening, enrollment, and trial onboarding tools. Unlike paper forms, researchers will not have to re-enter data collected by eConsent into the trial database before use.
And the benefits of using eConsent for a trial go beyond saving trees and time—eConsent can vastly improve recruitment and retainment efforts and streamline data flow.
For example, Castor’s eConsent tool helped Julius Clinical recruit over 17,825 participants in just 15 weeksfor their COVID-RED (Remote Early Detection) study COVID-RED provided participants with a wearable medical device and app that alerted them about a possible COVID-19 infection in real-time, so they could get tested before symptoms appeared. Travel restrictions and a high-risk subset of participants made onsite informed consent impossible.
Participants completed the enrollment process in the safety of their homes using Castor’s eConsent tool. eConsent data went directly to Castor’s electronic data capture (EDC) system so that participants could receive their wearables and get feedback as soon as possible.
This trial would not have been possible during a global pandemic without eConsent.
How to include eConsent in your IRB submission
If you plan on including eConsent at any point in your clinical trial, you must submit your plans to your IRB for approval. The IRB will want to see clear documentation detailing how eConsent supports participants’ decision-making during the consent process and throughout the trial. Developing your eConsent before submitting results in fewer requests for changes by the IRB—saving you time and money.
IRB members must engage with the same materials as participants to understand their effect. So they will need access to all the electronic material used in the eConsent tool, not just the content. They will consider interface usability, whether the material is accurate and suitable for participants, and the validity of hyperlinked information. Although IRB rules vary by locale, here are a few elements to consider as you prepare for submission:
Element #1: Consent workflow
Include whether the consent discussion will be face-to-face, remote, or a combination. If not face-to-face, the IRB will review whether the procedures effectively meet the goals of informed consent.
Questions to consider:
How will participants provide proof of identification?
How will participants be approached for consent? When will the question be asked?
What platform will host the informed consent discussion (for example, smartphone, video calls, Castor eConsent videoconferencing)?
Element #2: Patient comprehension assessment
An IRB submission must clearly explain how researchers will ensure patients meet the criteria of informed consent.
Consider:
How will the study be explained to participants?
How will their understanding be assessed (for example, popup quizzes)?
How will questions be answered?
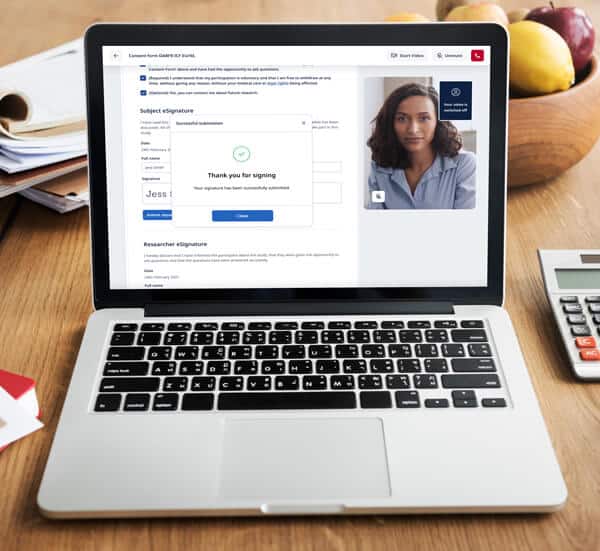
Element #3: eSignature process
Although an electronic signature (eSignature) is an integral aspect of eConsent, eSignature requirements vary by country, impacting global eConsent adoption. For example, Germany accepts both eConsent documents and eSignatures, while its neighbor France is wary of eConsent and does not allow eSignatures.
In the US, a valid eSignature can be the subject’s typed name or even a checkmark, as long as the symbol is logically associated with the person making it. Some software solutions, such as Castor’s eConsent, incorporate a valid eSignature in their platform.
Consider the following:
How will the electronic signature be created?
How can the signature be proven legitimate?
According to HIPAA regulatory requirements, eSignatures are only valid if participants can get an email or paper copy of the contract. In your application, consider how participants can get a copy of their consent (ex: download a pdf, receive an emailed form, or receive a paper form in the mail).
Element #4: Special circumstances
Finally, an IRB submission may need a section to address special circumstances related to eConsent per a trial’s unique needs.
The section might answer questions such as:
How will assent and parental permissions be obtained for minors?
Do participants need to be reconsented during the study?
Will caregivers take part in the study alongside participants?
How will the translation be addressed for non-English speakers? How will consent be witnessed in this scenario?
What if a participant doesn’t have the right technology to use eConsent?
For example, researchers can use LLM-generated video content that has passed human approval to engage participants in the informed consent process. Also, incorporating LLM chatbots in the eConsent process could answer patients’ questions in real time. Chatbots thoroughly trained in the trial protocol could replicate the onsite experience at home, reducing patient travel requirements and site burden.
The research community isn’t ready yet to incorporate patient-facing tools powered by LLMs, like chatbots, into the consent process. But LLMs hold the immediate potential to lighten the workload for trial staff and IRBs by generating content for human review and streamlining processes.
eConsent holds enormous potential for making informed consent more accessible for trial participants and simpler for sponsors and investigators. It also aligns with IRB efforts to ensure participants understand exactly what they are getting into when signing up for a trial. When preparing your submission, consider how the elements unique to eConsent, such as eSignatures and content delivery, will fit IRB requirements.
Considering eConsent for your clinical trial? The best time to evaluate how eConsent fits into your plans is before you submit your application to your IRB. Learn more about Castor’s eConsent platform and try it out today.
Comments closed
TRY CASTOR EDC FOR YOURSELF
Start designing your own study structure and forms today.